





Talasemitë janë një grup sëmundjesh gjenetike të trashëgueshme nga prindërit tek fëmijët dhe shkaktohen nga një defekt në prodhimin e hemoglobinës. Talasemitë bëjnë pjesë në grupin e anemive hemolitike mikrocitare.
Talasemitë karakterizohen nga defekte në prodhimin e vargjeve alfa ose beta globinike, që janë pjesë përbërëse të hemoglobinës (proteina e pranishme në qelizat e kuqe të gjakut që transporton oksigjenin). Rruazat e kuqe (eritrocitet) që prodhohen janë të varfra në hemoglobinë dhe shkatërrohen shpejt duke shkaktuar anemi (ulje të nivelit të hemoglobinës, hematokritit apo eritrociteve). Nëse kjo anemi nuk trajtohet, ndodhin ndryshime skeletike për shkak se palca e kockave ku prodhohen rruazat e kuqe (eritrocitet), rritet në vëllim duke u përpjekur të kompensojë humbjen e tyre. Rruazat e kuqe shkatërrohen shpejt (hemolizë) duke shkaktuar zmadhim te shpretkes (splenomegali), e më pas komplikacione të tjera, si anemia dhe grumbullim të hekurit në organizem.
Klasifikimi
Hemoglobina përbëhet nga katër zinxhirë proteinash, dy zinxhirë alfa globin dhe dy zinxhirë beta globin dhe në varësi të vargut që preket Talasemia ndahet në alfa talasemi dhe beta talasemi.

1-Alfa talasemia
Alfa-talasemia është veçanërisht e shpeshtë tek personat me origjinë nga Afrika e Veriut, Mesdheu ose Azia Juglindore.
Nga secili prind trashengohen tek fëmija 2 gjene, që krijojnë zinxhirë proteinash alfa globine. Kur një ose më shumë gjene janë me defekt ose mungojnë zhvillohet alfa talasema. Numri i gjeneve me defekt që trashëgohen do të përcaktojë nëse do të zhvillohen simptoma të anemisë dhe (nëse po) sa të rënda do të jenë ato.
A- Nëse është vetëm një gjen alfa me defekt ose mungon do të thotë që tek personi bartës nuk do të zhvillohet anemia. Kjo gjendje quhet “alfa talasemi minimale”.
B- Nëse janë dy gjene alfa me defekt ose mungojnë mund të zhvillohen simptoma të lehta të anemise (alfa talasemi minore).
C- Nëse janë tre gjene alfa të dëmtuara ose mungojnë zhvillohen simptoma të moderuara deri të rënda të anemisë (sëmundja e hemoglobinës H) HbH.
D- Nëse të katërta gjenet alfa janë të demtuar ose mungojnë zakonisht çojnë ne vdekje të fetusit ose të foshnjes direkt mbas lindjes (alfa talasemi majore ose Hydrops fetalis).
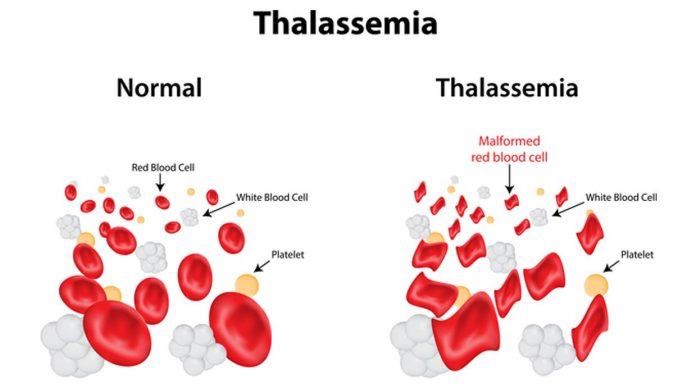
2-Beta talasemia
Beta-talasemia është më e shpeshtë tek personat me origjinë, Mesdhetare Lindja e Mesme, Azia Juglindore ose India.
Nga secili prind trashengohen tek fëmija 2 gjene, që krijojnë zinxhirë proteinash beta globine. Kur një ose më shumë gjene janë me defekt ose mungojnë zhvillohet beta talasema.
A- Nëse një gjen beta është i demtuar ose mungon mund të zhvillohen simptoma të lehta të anemisë (beta talasemi minore)
B- Beta talasemia intermedia është e karakterizuar nga simptoma te lehta deri në të moderuara të anesmise.
C- Beta Talasemia Major, talasemia madhore ose anemia e Cooley shkaktohet nga një mungesë e rëndë e beta globinës. Pacientët e prekur nga kjo formë zhvillojnë anemi të rëndë dhe hiperaktivitet të palces së kockave. Beta-talasemia major shfaqet brenda vitit të parë ose të dytë të jetës me simptoma të anemisë së rëndë dhe grumbullim të hekurit në organizem si pasojë e transfuzioneve të përsëritura dhe hemolizës. Grumbullimi i hekurit mund të shkaktojë dëmtim të funksionit të zemrës dhe siderozë hepatike me pasojë demtim të funksionit të melçisë deri në cirrozë hepatike.
Beta talasemia major mund të shkaktojë;
– Verdhëz
– Ulçera në këmbë
– Gurë në fshikëzën e tëmthit
– Splenomegali (zmadhim të shpretkës)
– Hiperplazi të palcës kockore që shkakton trashje të kockave të kafkës dhe eminencave malare. Përfshirja e kockave të gjata predispozon për fraktura patologjike dhe ngadalëson rritjen, duke vonuar ose bllokuar pubertetin.
Diagnoza
1-Analiza e gjakut periferik që konsiston në:
– Numërimin e plotë të rruazave të kuqe, që përfshinë masat e hemoglobinës, sasinë dhe madhësinë e qelizave të kuqe të gjakut. Personat e prekur nga talasemia kanë më pak qeliza të kuqe të gjakut të shëndetshme dhe më pak hemoglobinë se normalja. Ata gjithashtu mund të kenë qeliza të kuqe të gjakut më të vogla se normalja.
– Numërimin e retikulociteve (një masë e qelizave të reja të kuqe të gjakut) mund të tregojë se palca e kockave nuk prodhon mjaftueshëm qeliza të kuqe të gjakut
– Studimin e hekurit në gjak që tregon nëse shkaku i anemisë është mungesa e hekurit apo talasemia.
2-Elektroforeza e hemoglobinës: Tregon pranine e vargjeve globinike defektoze dhe /ose mungesen e vargjeve globinike.
3-Analiza e ADN-së :përdoret për diagnozën prenatale.
Trajtimi
Trajtimet baze për talaseminë major janë transfuzioni i gjakut (masa eritrocitare) dhe mjekimi me ferrokelante.
1-Transfuzioni i gjakut (masa eritrocitare), për të rivendosur nivelet normale të qelizave të kuqe të gjakut dhe hemoglobinës së shëndetshme.
2-Terapia me ferokelante, përdor medikamente të cilat lidhin dhe largojne hekurin e tepert prej organizmit. Një rrezik me transfuzionet e gjakut është se ato mund të shkaktojnë mbingarkesë hekuri në organizëm dhe kjo mund të dëmtojë organet. Terapia me ferrokelante orale (barna që lidhin dhe largojnë hekurin e tepert, si Deferasiroxi dhe Deferiproni) ka bërë një transformim spektakolar për të sëmurët me talasemi. Këto trajtime zgjasin për gjatë gjithë jetës.
3-Splenektomia, (heqje e shpretkes) nëse është e pranishme splenomegalia (zmadhim i shpretkës).
4-Transplantimi i qelizave staminale, qelizave burimore të palcës kockore për çdo pacient i jep mundësi mbi 90% të pacienteve për tu shëruar. Kjo në rastet e dhuruesve familjarë, të pajtueshëm sipas sistemit të HLA-së. Të dhëna inkurajuese ka edhe kur transplanti kryhet nga dhurues jo familjarë ose kur përdoren qelizat e kordonit umbilikal. Këto rezulatet janë shumë të kënaqshme kur ato kryhen sa më herët, që në 10 vjeçarin e parë të jetës.
5- Luspatercepti, është një injeksion që jepet çdo tre javë dhe stimulon palcën e kockave të prodhojë më shumë qeliza të kuqe të gjakut. Është miratuar në SHBA për trajtimin e beta talasemisë së varur nga transfuzioni.
Sigurisht për një ndjekje të mirë të pacientëve talasemikë, periodikisht bëhen kontrolle nga mjekë kardiologë, hepatologë, reumatologë, endokrinologë, infeksionistë, ortopedë, kirurgë, gjinekologë etj, duke ndërhyrë sipas rastit, apo komplikacioneve që ato paraqesin.
Parandalimi
Mund të parandalohet me anë të programeve parandaluese e depistuese, tek të gjithë të rinjtë, jo vetëm tek ciftet me rrezik te lartë.
Kryerja e Elektroforezës së Hemoglobinës, identifikon bartesit e Talasemisë, duke sjellë të dhëna për nivelin e HbA2, HbA, HbF.

Konsulta me mjekun pediatër hematolog, ndihmon në interpretimin e rezultatit të analizës. Nga ana tjetër, nëse një çift dëshiron të ketë femijë, dhe njëri apo të dy prindërit vuajnë nga Talasemia apo jane bartës, fillimisht ata duhet të informohen për rrezikun që femija i tyre mund të preket nga talasemia. Çifti duhet të informohet gjithashtu për mundësinë e diagnozës para lindjes. Kjo diagnozë mund të bëhet nëpërmjet biopsisë së vileve koriale, amniocentezës, kordocentezës apo me metoden e re te kuldocentezës.
Zgjedhja e llojit të testit varet nga faza e shtatzanisë dhe nga rezultatet e vlerësimit paraprak te prindërve.
Ekzaminimi i vileve koriale (biopsia), mund të kryhet në fazën e hershme të shtatzanisë, pas javës së 10-të. Merret një kampion i ADN-së nga placenta dhe analizohet për të parë praninë ose jo të mutacioneve gjenike.
Amniocenteza kryhet më vonë, pas javës së 15-të të shtatzanisë, duke marrë kampion nga lëngu amniotik, nën kontrollin e ekografisë.
Kuldocenteza është test i ri i diagnozës prenatale që jep rezultate të besueshme nga muaji i dytë i shtatzanisë, pra një muaj para vilocentezës. Kampioni i marrë përmban qeliza fetale, dhe ai merret nëpërmjet rrugës transvaginale pa patur nevojë për të shpuar qesen amniotike apo placentën, çka do të thotë që ka rrezik më të vogel për abort dhe për të shkaktuar keqformime fetale.
